Retinitis Pigmentosa, also known as RP, is a group of rare, inherited eye disorders that affect the retina, the light-sensitive cell layers at the back of the eye. It causes the cells in your retina to slowly break down, leading to severe visual impairment over time. Many adults with this eye disease experience significant vision loss.
Retinitis pigmentosa symptoms
The symptoms of retinitis pigmentosa typically start appearing in late childhood or adolescence and worsen over time, with the visual field narrowing in adulthood. It starts with night vision loss (nyctalopia) and progresses to peripheral (side) vision loss, eventually leading to complete vision loss.
By the age of 40, most people who have hereditary retinitis pigmentosa will have low vision and some might even become legally blind. However, this happens very slowly, with subtle symptoms, due to which it’s often overlooked.
Some of the most common symptoms of retinitis pigmentosa are:
- Night blindness: It’s when you find it difficult to see or move around in low light. In this eye condition, your eyes take longer to adjust to darkness. This includes environments like movie theatres and dimly lit rooms, as well as situations like trouble driving at night or in the dusk.
- Blind spots in your peripheral vision: Also known as tunnel vision, it happens when you are unable to see objects out of the corners of your eyes, eventually narrowing your field of vision. This means you might bump into things around you.
- Central vision loss: This can make it difficult to perform close-up, detailed tasks such as reading, threading a needle or recognising approaching faces.
- Colour vision deficiency: This means it becomes difficult for you to distinguish between different colours.
- Light sensitivity: Also known as photophobia, this happens when someone becomes highly sensitive to bright light

Causes of retinitis pigmentosa
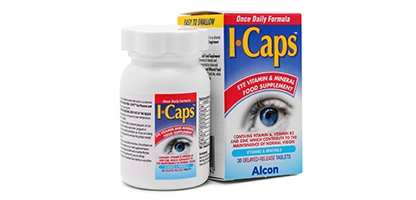
Your retina converts light into electrical signals, which your brain interprets as images, giving you the ability to see. As photoreceptor (light-sensing) cells in the retina start degenerating, hereditary retinitis pigmentosa changes the retina’s response to light, eventually making it difficult to see.
Wut Win, a Dispensing Optician at Feel Good Contacts, explains, “RP is caused by genetics, which means you can inherit it from a biological parent or have a spontaneous mutation.”
Your genes play a major role in building your cells, controlling cells in your retina and operating your body. Research indicates that there are nearly 100 genetic variations that can cause RP through different pathways. This is why scientists consider it a group of eye disorders rather than a single disorder. It’s also why different people might have different types of vision loss at varying levels of severity, depending on their condition. Some people might develop this eye disease as part of a bigger genetic condition that affects several organs, such as Usher syndrome (it leads to both hearing and vision loss).
Some external factors including certain infections, medications or an eye injury can cause pseudo-retinitis pigmentosa.
Treatment for retinitis pigmentosa
There is no cure for retinitis pigmentosa. However, vision rehabilitation (training) programs and vision aids can make it easier to live with low vision. These include:
- Support or counselling groups
- Vocational or special education services
- Occupational therapy
- Assistive devices or visual aids
You can consult an eye doctor who can suggest some eye vitamins and supplements for RP, such as Eyetamins Vision Support and ICaps Tablets.

 Need Optical Advice?
Need Optical Advice?

 Offers
Offers Account
Account
 Menu
Menu Favorite
Favorite
 Basket
Basket
 Offers
Offers